La osteogénesis imperfecta también se produce por mutaciones en ‘BMP1’, que dificultan la formación de colágeno I funcionalmente maduro. Este nuevo hallazgo, en el que ha participado el CSIC, mejorará la capacidad de diagnóstico de dicha enfermedad rara.
La osteogénesis imperfecta también se produce por mutaciones en ‘BMP1’, que dificultan la formación de colágeno I funcionalmente maduro. Este nuevo hallazgo, en el que ha participado el CSIC, mejorará la capacidad de diagnóstico de dicha enfermedad rara.
Versiones mutadas del gen BMP1 son una causa más de la osteogénesis imperfecta, según una investigación liderada por un investigador del Consejo Superior de Investigaciones Científicas (CSIC).
Este gen codifica una enzima encargada de cortar una parte de la cadena proteica de las moléculas de procolágeno I que posteriormente se convierten en moléculas de colágeno I funcionalmente maduras. La mutación descrita en este trabajo provoca que los BMP1 modificados no realicen este proceso de forma correcta.
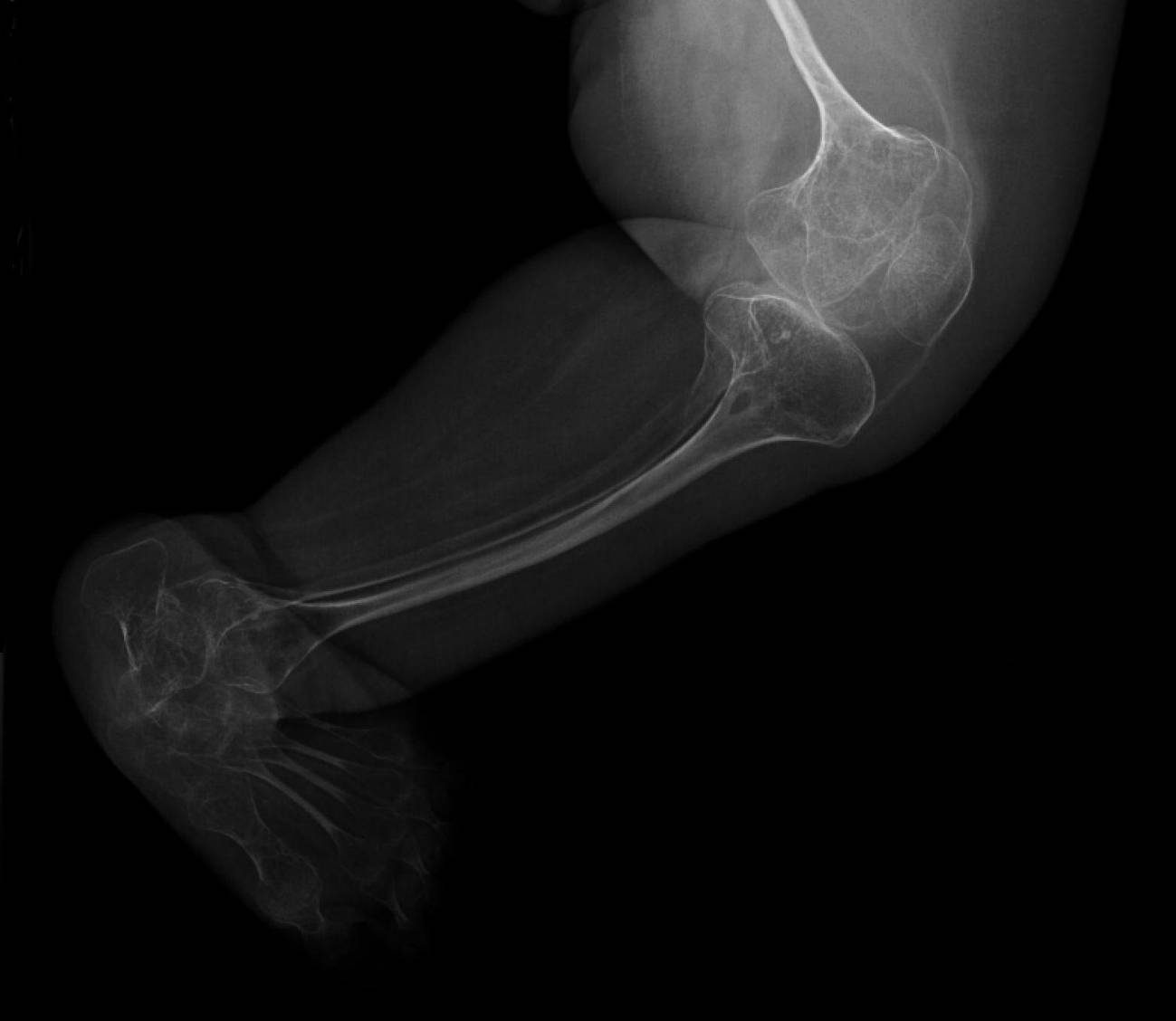
Esta enfermedad rara, comúnmente conocida como huesos de cristal, se caracteriza por una deficiencia en la producción de colágeno I, lo que provoca una fragilidad excesiva en los huesos. Dicha dolencia afecta, aproximadamente, a unas 2.700 personas en España.
La variante dominante de esta enfermedad rara, que es la más común, se caracteriza por mutaciones en los genes COL1A1 y COL1A2. También existe una minoría de casos de herencia recesiva en los que, durante los últimos años, se han descrito mutaciones en un número cada vez mayor de genes.
Nuevo gen a tener en cuenta
“Este es un nuevo gen a tener en cuenta en el diagnóstico genético de esta patología”, explica Víctor Luis Ruiz‐Pérez, líder de la investigación e investigador del Instituto de Investigaciones Biomédicas Alberto Sols (centro mixto del CSIC y la Universidad Autónoma de Madrid).
Los resultados, publicados en la revista Human Mutation, se basan en el estudio de una familia egipcia con dos niños afectados por una variante recesiva y grave de osteogénesis imperfecta.
En 2010, este mismo equipo descubrió la implicación del gen OSTERIX/SP7 en el desarrollo de la enfermedad. Los resultados se publicaron en la revista The American Journal of Human Genetics.
La investigación también ha contado con la colaboración del equipo de investigación de Pablo Lapunzina del Hospital Universitario La Paz de Madrid que junto al centro de Ruiz‐Pérez integran el Centro de Investigación Biomédica en Red de Enfermedades Raras.
Referencia bibliográfica
Víctor Martínez González, María Valencia, José A. Caparrós Martín, Mona Aglan, Samia Temtamy, Jair Tenorio, Verónica Pulido, Uschi Lindert, Marianne Rohrbach, David Eyre, Cecilia Giunta, Pablo Lapunzina, Víctor L. Ruiz Pérez. “Identification of a mutation causing deficient BMP1/mTLD proteolytic activity in autosomal recessive osteogenesis imperfect”. Human Mutation. Doi: 10.1002/humu.21647

Un análisis de datos de más de 2,5 millones de personas identifica 26 regiones del genoma asociadas a este síndrome de dolor crónico. Los resultados señalan posibles dianas terapéuticas, pero todavía no permiten diagnosticar la enfermedad mediante una prueba genética ni se traducen en nuevos tratamientos.

Un equipo internacional con participación española ha observado por primera vez un mecanismo de ‘cápsula de escape’ en comunidades bacterianas que anteriormente se habían relacionado con animales marinos. El objetivo de este fenómeno es garantizar que la comunidad sobreviva en nuevas colonias cuando se detecta una amenaza.

